Mechanochemistry has often been heralded as the next frontier in chemistry, where mechanical forces rather than traditional chemical reagents drive molecular transformations. Among the most promising contributors to this field are mechanophores—molecules engineered to physically or chemically respond when subjected to mechanical stress. A remarkable stride has just been made in understanding how certain critical chemical bonds, specifically carbon-carbon (C–C) bonds, respond to tension. This advancement promises to transform how new materials and therapeutic agents are designed, offering new avenues for innovation in material science, organic synthesis, and medicine.
From Complexity to Clarity: The Challenge of C–C Bond Reactivity
One of the toughest dilemmas in mechanochemistry is predicting the exact conditions under which a C–C bond will break under mechanical force. Until now, this prediction demanded extensive computational resources and laborious experimental validation given the complex nature of forces applied in diverse directions—pulling, pushing, or shearing. The inherently unpredictable “vectoral” characteristics of these forces meant researchers were often navigating blind, relying on trial and error to understand how molecules like mechanophores would behave.
However, researchers led by Prof. Jeffrey Moore and graduate student Yunyan Sun, collaborating with teams from MIT and Duke University, have introduced a refreshing simplification to this analytical paralysis. Their newly developed computational tool, described in their recent publication in Chem, promises to forecast the mechanochemical reactivity of C–C bonds with reliable accuracy while bypassing exhaustive calculations.
The Tension Model of Bond Activation: A Fresh Perspective
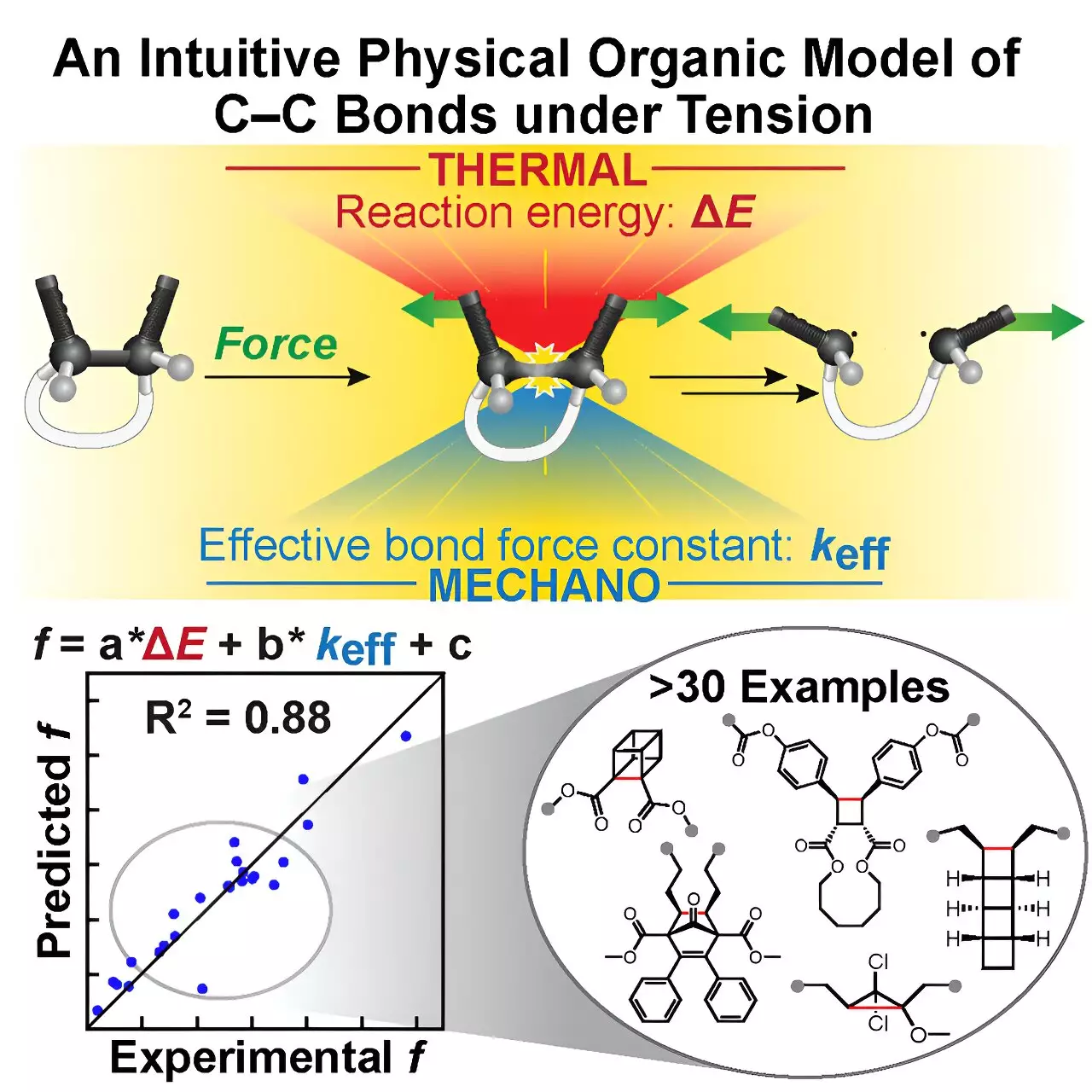
Dubbed the Tension Model of Bond Activation (TMBA), this tool is grounded in a simple yet powerful concept derived from the Morse Potential— a well-known classical chemistry model used to describe bond energy. TMBA leans on two accessible molecular parameters: the effective force constant and the reaction energy, to evaluate the bond’s behavior under stress.
The genius of TMBA lies in the “restoring force triangle,” a conceptual framework that unifies these parameters into a comprehensible geometric visualization. This triangle enables chemists to intuitively grasp how force applied to a molecule translates into bond activation—turning what was once an abstract computational output into an immediate, interpretable insight. This democratization of mechanochemical prediction is a significant leap toward making advanced molecular design approachable, even at the undergraduate level.
Why Intuitive Models Matter More than Ever
Modern chemistry has long benefited from simplified mental models to teach and rationalize complex reactions—from the Lewis structures to Marcus theory for electron transfer. TMBA aligns perfectly with this tradition. It bridges the formidable gap between abstract quantum mechanics-level calculations and practical experimental chemistry, offering a scaffold that supports both novice learners and seasoned researchers.
Graduate researcher Yunyan Sun emphasizes the importance of an intuitive tool that can guide chemists without overwhelming them with data and computational jargon. High-level simulations are invaluable but often inaccessible or impractical at early stages of mechanophore design. TMBA fills this gap by offering a “first look” at the mechanochemical landscape, enabling researchers to predict and refine molecular behavior before engaging in demanding computational workloads.
Collaboration at the Core of Innovation
The development of TMBA vividly illustrates the power of interdisciplinary and inter-institutional collaboration. Contributions from Duke’s Stephen L. Craig and his team added experimental single-molecule studies that validated the computational model, while MIT’s chemical engineering expertise was integral in refining the theoretical framework. This convergence of specialized knowledge within MONET (Center for the Chemistry of Molecularly Optimized NETworks) demonstrates how collaborative synergy can dismantle scientific bottlenecks.
Such cooperation also accelerated the revelation of a generalizability in mechanophore reactivity trends, moving beyond isolated case studies to broader principles applicable across various molecules. This generalization not only enhances predictive power but also suggests that the model could underpin the discovery of entirely new mechanochemical reactions and materials.
Implications: Teaching, Therapeutics, and Tomorrow’s Materials
TMBA’s straightforward, teachable nature makes it poised to impact not just research labs but also the academic curriculum. Envision a future chemistry course where freshmen grasp the dynamic reactivity of C–C bonds under mechanical stress—a concept once reserved for advanced researchers. Breaking the misconception that these bonds are indestructible under ordinary conditions opens a world of possibilities.
Moreover, the ability to precisely control and predict mechanophore activation has direct biomedical relevance. The previously developed mechanophore known as NEO, which can mechanically release controlled doses of carbon monoxide—a molecule with therapeutic potential—exemplifies this promise. Such precision-controlled drug delivery methods could revolutionize treatments by minimizing side effects and improving targeting.
A Paradigm Shift in Mechanochemical Science
This work signifies a pivotal shift from an era dominated by painstaking trial and error to one buoyed by rational design enabled through elegant simplicity. Mechanochemistry, once a niche with unpredictable outcomes, now stands on firmer theoretical ground thanks to TMBA. The model challenges entrenched ideas about bond resilience and opens up a playground where mechanical forces become precise tools for innovation.
In the broader scientific landscape, such advances underscore the value of clarity and accessibility in complex fields. The researchers’ success with TMBA should inspire similar efforts to distill intricate phenomena into user-friendly models that inform and accelerate discovery, fostering progress across the sciences.