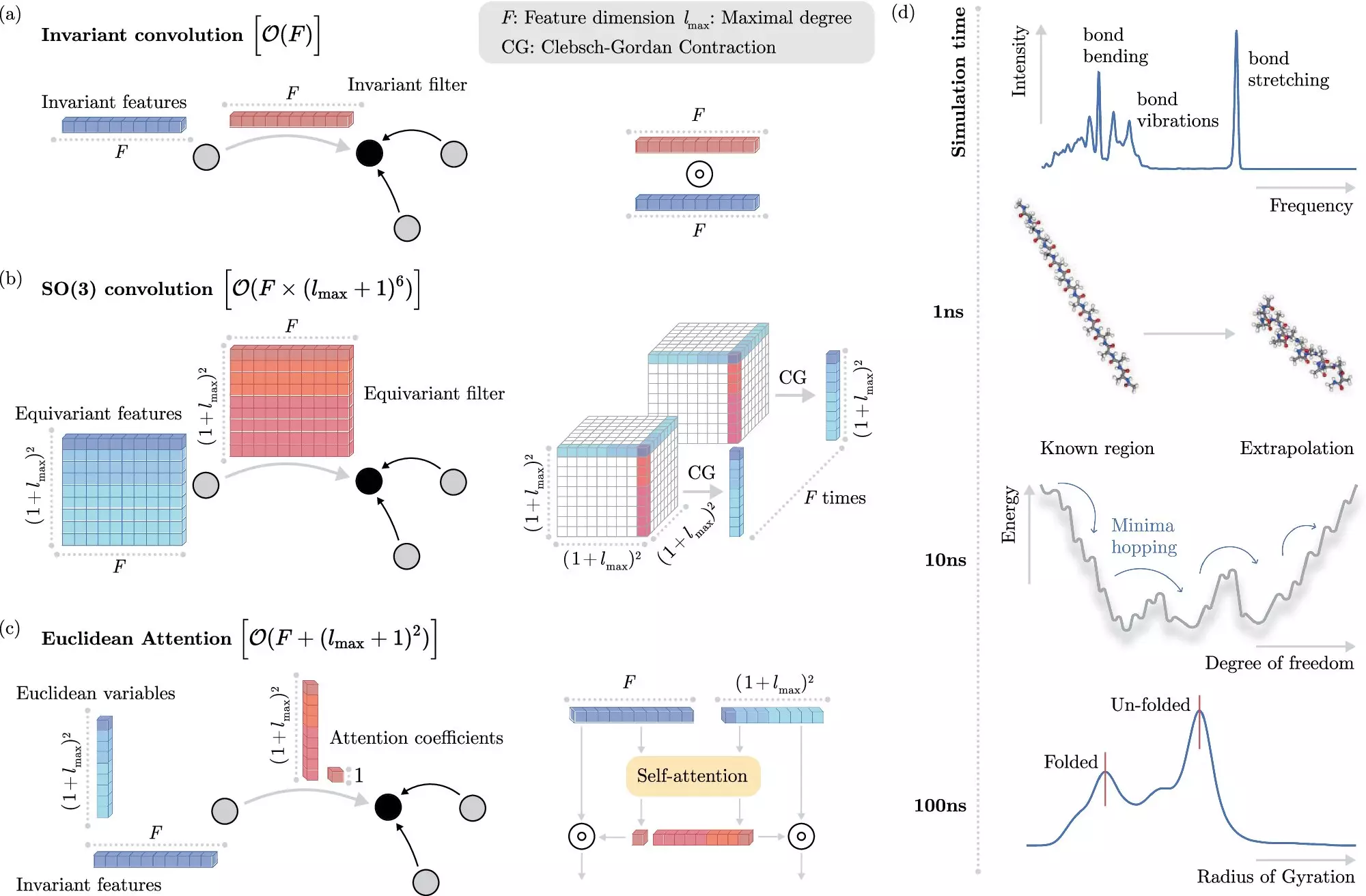
Molecular dynamics simulation, at its core, is the gateway to understanding the behavior of matter at the molecular level. Atoms, which serve as the fundamental building blocks of the universe, engage in complex interactions shaped by their quantum mechanical nature. Each atom is made up of a positively charged nucleus surrounded by a swarm of negatively charged electrons. The task of simulating how these atoms bond to form molecules, and the behavior those molecules exhibit, is breathtakingly intricate. Traditional methods face simmering limitations, particularly when it comes to scenarios involving multiple atoms and prolonged time scales, leading to arduous computations taking days or even longer on high-powered computing systems.
The cornerstone of many classical computational methods lies in solving the Schrödinger equation—an essential mathematical framework for quantum mechanics. Unfortunately, obtaining precise solutions for molecules that contain more than a few dozen atoms is rarely swift. As the number of atoms increases, so too does the time and complexity involved. Solving this equation multiple times—sometimes thousands or millions, particularly for simulations over extended periods—exposes critical flaws in conventional approaches, making them increasingly untenable in modern scientific research.
Machine Learning: The Game Changer
In recent years, however, breakthroughs in machine learning (ML) have offered a promising alternative to these age-old problems. ML systems, unlike traditional methods, can sidestep the complexities of directly solving the Schrödinger equation by learning to predict electronic interactions effectively. The advantage lies in their ability to do this at a fraction of the computational cost, provided their algorithms are accurately tuned.
One of the most pressing challenges in applying ML to molecular simulations involves understanding how electrons interact. Traditional ML models process vast amounts of input data but often fall prey to inefficiencies when seeking to impose physical invariances—the fundamental properties of molecules that hold constant as their relative positions fluctuate. Addressing these computational hurdles without losing fidelity has been a significant roadblock in the deployment of ML in complex quantum systems.
A Breakthrough from BIFOLD and Google DeepMind
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) in collaboration with Google DeepMind have recently made groundbreaking strides in overcoming these barriers. Their novel ML algorithm, featured in the esteemed journal Nature Communications, represents a significant leap in simulating molecular dynamics with exceptional accuracy and reduced computational expense.
By innovatively decoupling the learning algorithms from the complexity of modeling individual electron interactions, their approach alleviates the computational burdens faced by traditional methods. This modification enables simulations that would have taken months or years to complete on high-performance computing clusters to be performed in a matter of days—an efficiency leap that transforms the landscape of computational chemistry.
Applications: Drug Development and Beyond
In practical terms, these advanced simulations could have vast implications for scientific fields like drug discovery and material design. Imagine a future where the precise modeling of molecular interactions allows researchers to develop new drugs with unprecedented efficiency and environmental consciousness, fundamentally altering our approach to pharmaceutical research. The potential for accelerated experimentation is particularly timely in a world increasingly driven toward sustainable practices.
For instance, the research team utilized their new ML techniques to analyze docosahexaenoic acid (DHA), an essential fatty acid critical to brain functions and development. This fine-tuned ML method could pinpoint the most stable version of DHA by sifting through tens of thousands of potential molecular candidates with precision, something traditional methods would deem prohibitive.
Pushing the Boundaries of Computational Chemistry
The synergy between advanced machine learning techniques and physical principles not only enriches the discourse surrounding computational chemistry but also continues a vital line of research. Future generations of these algorithms need to be adept at capturing more intricate interactions, honing in on complex, long-range physical behaviors that are essential for a true depiction of diverse chemical systems.
As the insights garnered from these advancements unravel further mysteries of nature at the atomic scale, they pave the way for a new era of research—one that promises to deepen our understanding of molecular interactions and opens our eyes to possibilities both unfathomed and exciting. The ongoing collaboration between cutting-edge research institutions and tech giants like Google signals a promising frontier, where the power of AI converges with the foundational sciences to unlock realms of knowledge that were once relegated to the realm of dreams.